Đây là công cụ truyền thống nhưng được ứng dụng rộng rãi và có nhiều vai trò quan trọng trong nghiên cứu protein. Thứ nhất chúng làm cho các mẫu phức tạp trở nên đơn giản hơn bằng cách phân tách hỗn hợp mẫu thành các protein riêng lẻ hoặc các nhóm nhỏ. Thứ hai, chúng cho phép phân biệt sự khác nhau về mức độ biểu hiện của protein, và so sánh giữa các mẫu. Có 5 kỹ thuật chính để phân tách protein đó là điện di 1 chiều SDS-PAGE, điện di 2-DE, điện di đẳng điện IEF, sắc ký lỏng nano hiệu năng cao HPLC, ngoài ra còn nhiều phương pháp phân tách khác nhau tùy thuộc vào mục đích và nhu cầu sử dụng.
Điện di SDS-PAGE
SDS-PAGE được thực hiện theo LaemLi [71]. Trong phương pháp này, các phân tử protein được phân tách theo trọng lượng dưới tác dụng của điện trường không đổi. Protein được phân tách trong gel polyacrylamide với các nồng độ khác nhau. Dưới tác dụng của dòng điện một chiều, các protein có kích thước khác nhau sẽ di chuyển về điện cực trái dấu. Các phân tử protein gắn với SDS nên chúng sẽ tích điện âm, do đó sự khác biệt về điện tích được loại trừ.
Điện di SDS-PAGE
SDS-PAGE được thực hiện theo LaemLi [71]. Trong phương pháp này, các phân tử protein được phân tách theo trọng lượng dưới tác dụng của điện trường không đổi. Protein được phân tách trong gel polyacrylamide với các nồng độ khác nhau. Dưới tác dụng của dòng điện một chiều, các protein có kích thước khác nhau sẽ di chuyển về điện cực trái dấu. Các phân tử protein gắn với SDS nên chúng sẽ tích điện âm, do đó sự khác biệt về điện tích được loại trừ.
.png)
Khi protein được chạy trong điện trường không đổi, phức hệ protein-SDS sẽ di chuyển xuyên qua các lỗ gel polyacrylamid với vận tốc phục thuộc vào hình dáng, kích thước phân tử. Khi đó protein sẽ được phân tách thành các băng, vạch khác nhau. Gel thường được sử dụng với nồng độ từ 5%-15% và có thể được chạy theo chiều nằm ngang hoặc chiều thẳng đứng (Hình 5) [40].
Điện di 2-DE
Ngày này, kỹ thuật điện di 2 chiều (two-dimensional electrophoresis, 2DE) ngày càng trở thành một công cụ được ứng dụng rộng rãi nhằm phân tách các phức hợp protein trong tế bào, mô và các dịch cơ thể [46]. Phương pháp điện di hai chiều, thủy phân protein trong gel, sau đó nhận dạng bằng khối phổ đang là một phương pháp truyền thống hay được sử dụng nhất. Tùy thuộc vào kích thước lỗ gel và giải gradient pH sử dụng, điện di 2-DE có thể phân tách hàng ngàn protein (5000 protein, và có thể phát hiện vệt protein với lượng <1ng) có mặt trong mẫu cùng một lúc, đồng thời so sánh mức độ biểu hiện khác nhau của các mẫu phân tích. Đây là phương pháp cổ điển nhưng vẫn được áp dụng rộng rãi và thường gắn liền với proteomics vì nó có khả năng phân tách cao, hiển thị hình ảnh so sánh tương quan, đem lại bức tranh toàn cảnh về mức độ biểu hiện khác nhau của các protein.
Điện di 2-DE
Ngày này, kỹ thuật điện di 2 chiều (two-dimensional electrophoresis, 2DE) ngày càng trở thành một công cụ được ứng dụng rộng rãi nhằm phân tách các phức hợp protein trong tế bào, mô và các dịch cơ thể [46]. Phương pháp điện di hai chiều, thủy phân protein trong gel, sau đó nhận dạng bằng khối phổ đang là một phương pháp truyền thống hay được sử dụng nhất. Tùy thuộc vào kích thước lỗ gel và giải gradient pH sử dụng, điện di 2-DE có thể phân tách hàng ngàn protein (5000 protein, và có thể phát hiện vệt protein với lượng <1ng) có mặt trong mẫu cùng một lúc, đồng thời so sánh mức độ biểu hiện khác nhau của các mẫu phân tích. Đây là phương pháp cổ điển nhưng vẫn được áp dụng rộng rãi và thường gắn liền với proteomics vì nó có khả năng phân tách cao, hiển thị hình ảnh so sánh tương quan, đem lại bức tranh toàn cảnh về mức độ biểu hiện khác nhau của các protein.

Kỹ thuật điện di 2-DE thực ra là sự kết hợp của hai nguyên lý phân tách khác nhau. Theo chiều thứ nhất, các phân tử protein được phân tách dựa theo điểm đẳng điện pI bởi nguyên lý hội tụ theo điểm đẳng điện IEF (Isoelectric focusing) trên một thanh strip có giải gradient pH xác định (ví dụ dải pH 3-10). Chiều thứ hai, các phân tử protein được phân tách trên điện di trên gel polyacrylamid (như điện di SDS-PAGE) (hình 6). Do đó, thực chất 2-DE là phương pháp phân tách protein theo 2 chiều, (i) chiều thứ nhất theo điện tích và (ii) chiều thứ hai theo trọng lượng phân tử.
Sự khác biệt về điểm đẳng điện pI (Isoelectric ponit) của các protein chính là cơ sở của IEF. pI là giá trị pH tại đó điện tích bề mặt tổng số của protein bằng 0 (protein không di chuyển trong điện trường). Mỗi protein có một giá trị pI xác định. Thanh gel IPG (immobilized pH gradient) tạo ra một giải gradient pH cố định, liên tục và tăng dần với chiều dài khác nhau (7 cm, 11 cm, 18 cm, 24 cm) đã hạn chế sự di chuyển, trôi dạt của các hóa chất (vì chúng được gắn trên gel polyacrylamid) làm cho quá trình phân tách ổn định hơn [84]. Ngoài điện di 2DE các kỹ thuật khác như điện di mao quản, điện di trường xung cũng thường được sử dụng để phân tách protein
Sắc ký lỏng nano đa chiều (2DnanoLC)
Mặc dù 2-DE được sử dụng rộng rãi như một phương pháp truyền thống để phân tách protein nhưng hạn chế của nó là độ nhạy thấp, không tự động, mất thời gian, tốn kém, tiến hành nhiều lần tạo ra sai số. Do đó phương pháp này gặp nhiều khó khăn trong quá trình phân tích các protein có hàm lượng thấp hoặc các protein có tính kỵ nước cao (như protein màng). Để vượt qua những hạn chế này sắc ký lỏng nano đa chiều (multidimensional nano liquid chromatography - MDnanoLC) được pháp triển như là một phương pháp thay thế hiệu quả (hình 8) [87].
Sự khác biệt về điểm đẳng điện pI (Isoelectric ponit) của các protein chính là cơ sở của IEF. pI là giá trị pH tại đó điện tích bề mặt tổng số của protein bằng 0 (protein không di chuyển trong điện trường). Mỗi protein có một giá trị pI xác định. Thanh gel IPG (immobilized pH gradient) tạo ra một giải gradient pH cố định, liên tục và tăng dần với chiều dài khác nhau (7 cm, 11 cm, 18 cm, 24 cm) đã hạn chế sự di chuyển, trôi dạt của các hóa chất (vì chúng được gắn trên gel polyacrylamid) làm cho quá trình phân tách ổn định hơn [84]. Ngoài điện di 2DE các kỹ thuật khác như điện di mao quản, điện di trường xung cũng thường được sử dụng để phân tách protein
Sắc ký lỏng nano đa chiều (2DnanoLC)
Mặc dù 2-DE được sử dụng rộng rãi như một phương pháp truyền thống để phân tách protein nhưng hạn chế của nó là độ nhạy thấp, không tự động, mất thời gian, tốn kém, tiến hành nhiều lần tạo ra sai số. Do đó phương pháp này gặp nhiều khó khăn trong quá trình phân tích các protein có hàm lượng thấp hoặc các protein có tính kỵ nước cao (như protein màng). Để vượt qua những hạn chế này sắc ký lỏng nano đa chiều (multidimensional nano liquid chromatography - MDnanoLC) được pháp triển như là một phương pháp thay thế hiệu quả (hình 8) [87].

Bằng sự kết hợp của các kỹ thuật phân tách các cột liên tiếp nhau dựa vào sự kết hợp của phương pháp sắc ký trao đổi ion (SCX), sắc ký ái lực và phương pháp sắc ký ngược pha (RP) [121]. Điểm đáng lưu ý trong phương pháp này, đó là kích thước cột rất bé (ID vài mm), và tốc độ dòng rất thấp (0,2 ml/phút).
Phương pháp sắc ký lỏng nano đa chiều kết nối trực tuyến với hệ khối khối QSTAR XL sử dụng các chiến lược cột sắc ký nhiều chiều khác nhau với đường kính cột và tốc độ dòng cực thấp đã và đang được sử dụng mạnh mẽ để phân tách và nhận diện các mẫu phức tạp [7]. Điểm quan trọng của phương pháp này đó là có thể sử dụng lượng mẫu nhỏ, tự động, chính xác và có khả năng phân tách tốt nhiều peptid cùng lúc chỉ trong một lần chạy [120]. Ngoài sắc ký lỏng nano, các phương pháp khác như sắc ký hiệu năng cao HPLC, sắc ký ái lực, sắc ký ngược pha cũng được sử dụng kết hợp để phân tách protein. Sự kết hợp này được gọi là sắc ký liên tục (tandem LC).
Khối phổ - Trung tâm của proteomics
Phương pháp khối phổ (Mass Spectrometry-MS) ngày càng được lựa chọn để phân tích các phức hợp protein, là một kỹ thuật được ứng dụng rộng rãi nhất và không thể thay thế trong các nghiên cứu hỗn hợp protein những năm gần đây [7, 9, 18, 30]. Proteomics dựa trên nguyên lý khối phổ đã trở thành một môn khoa học thực sự nhờ có các dữ liệu về trình tự gen và genome cùng với các thành tựu vượt bậc trong nhiều lĩnh vực [2]. Việc phát triển các thiết bị khối phổ có độ phân giải, độ nhạy, độ chính xác và tự động ngày càng cao làm cho khối phổ trở thành trung tâm trong nghiên cứu protein và lĩnh vực proteomics trở nên kỳ thú hơn. Các kỹ thuật này ngày càng không thể thay thế nhằm giải thích những thông tin được mã hoá trong genome.
Nguyên lý của khối phổ
Phương pháp khối phổ (Mass Spectrometry-MS) ngày càng được lựa chọn để phân tích các phức hợp protein, là một kỹ thuật được ứng dụng rộng rãi nhất và không thể thay thế trong các nghiên cứu hỗn hợp protein những năm gần đây [7, 9, 18, 30]. Proteomics dựa trên nguyên lý khối phổ đã trở thành một môn khoa học thực sự nhờ có các dữ liệu về trình tự gen và genome cùng với các thành tựu vượt bậc trong nhiều lĩnh vực [2]. Việc phát triển các thiết bị khối phổ có độ phân giải, độ nhạy, độ chính xác và tự động ngày càng cao làm cho khối phổ trở thành trung tâm trong nghiên cứu protein và lĩnh vực proteomics trở nên kỳ thú hơn. Các kỹ thuật này ngày càng không thể thay thế nhằm giải thích những thông tin được mã hoá trong genome.
Khối phổ là kỹ thuật phân tích đo phổ về khối lượng của các phân tử tích điện khi chúng di chuyển trong điện trường. Mẫu được ion hóa trở thành các phân tử tích điện khác nhau và được phân tách dựa vào sự sai khác về giá trị m/z. Dữ liệu phổ khối được tự động ghi lại và sử dụng để nhận dạng protein bằng các công cụ tin sinh học.
Hoạt động và cấu tạo chung của máy khối phổ
Ban đầu, mẫu (i) được ion hóa thành các ion ở các trạng thái tích điện khác nhau, (ii) sau đó các ion mẫu sẽ được phân tách dựa trên sự khác biệt về giá trị m/z, (iii) ghi dữ liệu phổ về khối của các ion đã phân tách với sự trợ giúp của các phần mềm tin sinh học cho phép tìm kiếm trên các cơ sở dữ liệu và phân tích kết quả thu được. Máy khối phổ không đo khối lượng (m) mà chúng đo giá trị m/z (mass/charge ratio).
Hoạt động và cấu tạo chung của máy khối phổ
Ban đầu, mẫu (i) được ion hóa thành các ion ở các trạng thái tích điện khác nhau, (ii) sau đó các ion mẫu sẽ được phân tách dựa trên sự khác biệt về giá trị m/z, (iii) ghi dữ liệu phổ về khối của các ion đã phân tách với sự trợ giúp của các phần mềm tin sinh học cho phép tìm kiếm trên các cơ sở dữ liệu và phân tích kết quả thu được. Máy khối phổ không đo khối lượng (m) mà chúng đo giá trị m/z (mass/charge ratio).
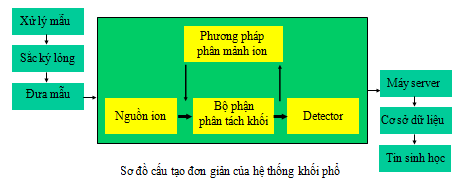
Hệ thống khối phổ có nhiều loại và được ứng dụng rất đa dạng trong các lĩnh vực khác nhau. Trong đó, có hai loại hệ thống hay được sử dụng chính trong proteomics là MALDI-TOF và ESI-MS/MS [2, 10]. Hai hệ thống này khá khác nhau nhưng bổ sung cho nhau với cùng một mục đích là nhận dạng protein. Máy khối phổ có 3 phộ phận chính đó là (i) nguồn ion hóa mẫu, (ii) bộ phận phân tích khối, (iii) bộ phận ghi phổ (detector). Tùy thuộc vào các loại nguồn và bộ phân tích khối mà máy khối phổ có cấu hình và nguyên tắc hoạt động khác nhau. Hình 9 minh họa sơ đồ cấu tạo đơn giản của hệ thống khối phổ.
Có 2 loại nguồn ion hóa mẫu hay được sử dụng đó là nguồn MALDI và ESI.
Kỹ thuật MALDI-TOF
MALDI là thuật ngữ chỉ phương pháp ion hóa mẫu hấp thụ dựa trên sự hỗ trợ của các chất nền và năng lượng laser (Matrix-assisted laser desorption ionization, MALDI). Nguồn MALDI sử dụng các chất nền hỗ trợ để ion hóa mẫu dưới tác dụng của năng lượng laser lớn.
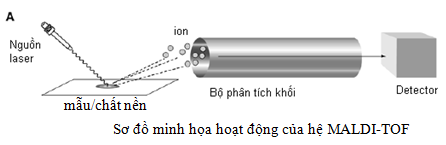
Ban đầu, mẫu được xử lý, làm tinh sạch bằng nhiều phương pháp khác nhau. Sau đó mẫu phân tích được trộn với các chất nền (các axit yếu như sinapinic) chứa các phân tử hữu cơ nhỏ có khả năng hấp thụ năng lượng ánh sáng ở những bước sóng nhất định. Hỗn hợp mẫu và chất nền được đưa lên khay và bay hơi tạo thành các hạt tinh thể bám với peptid. Dưới tác dụng của nguồn năng lượng laser cực lớn chiếu vào làm cho các chất nền (axit yếu) hấp thụ năng lượng và bật ra các photon. Mẫu được hấp thụ photon và năng lượng sẽ được đi vào hệ thống khối phổ TOF. Các ion dương thường được tạo ra đối với mẫu dạng các peptid/protein. Mỗi peptid có xu hướng hấp thụ một photon kết quả là ion peptid sẽ tích điện dương +1.
Phương pháp MALDI-TOF và MALDI-TOF MS thường được sử dụng cho các mẫu protein đơn giản, và khá tinh sạch [10]. Ưu điểm là có thể giải trình tự axit amin mảnh peptid và đo chính xác khối lượng, nhưng nó lại không thích hợp cho việc phân tích hỗn hợp protein phức tạp.
Kỹ thuật ESI-MS/MS
ESI (ElectroSpray Ionization) là thuật ngữ chỉ phương pháp ion hóa mẫu bằng phương pháp phun chùm ion trong dung dịch tạo thành đám sương mù với các giọt nhỏ dễ bay hơi.
Cơ chế hoạt động của nguồn ESI khá đơn giản. Dưới áp lực dòng liên tục, đường kính cột bé và hiệu điện thế cao (2500V), peptid sẽ được phun tơi thành các giọt nhỏ đa điện tích ra khỏi đầu kim phun. Kim phun sẽ tạo thành các giọt nhỏ, như đám sương mù (đám mây khí ion), dễ bay hơi, các giọt này chứa peptid và các thành phần dung môi khác. Quá trình bay hơi được thực hiện bởi nhiệt độ hoặc màng chắn khí nitơ (curtain gas) làm cho mẫu dễ bay hơi. Kết quả là chỉ còn peptid tích điện và bay vào bộ phận phân tích khối của máy khối phổ liên tục MS/MS.
Phương pháp MALDI-TOF và MALDI-TOF MS thường được sử dụng cho các mẫu protein đơn giản, và khá tinh sạch [10]. Ưu điểm là có thể giải trình tự axit amin mảnh peptid và đo chính xác khối lượng, nhưng nó lại không thích hợp cho việc phân tích hỗn hợp protein phức tạp.
Kỹ thuật ESI-MS/MS
ESI (ElectroSpray Ionization) là thuật ngữ chỉ phương pháp ion hóa mẫu bằng phương pháp phun chùm ion trong dung dịch tạo thành đám sương mù với các giọt nhỏ dễ bay hơi.
Cơ chế hoạt động của nguồn ESI khá đơn giản. Dưới áp lực dòng liên tục, đường kính cột bé và hiệu điện thế cao (2500V), peptid sẽ được phun tơi thành các giọt nhỏ đa điện tích ra khỏi đầu kim phun. Kim phun sẽ tạo thành các giọt nhỏ, như đám sương mù (đám mây khí ion), dễ bay hơi, các giọt này chứa peptid và các thành phần dung môi khác. Quá trình bay hơi được thực hiện bởi nhiệt độ hoặc màng chắn khí nitơ (curtain gas) làm cho mẫu dễ bay hơi. Kết quả là chỉ còn peptid tích điện và bay vào bộ phận phân tích khối của máy khối phổ liên tục MS/MS.
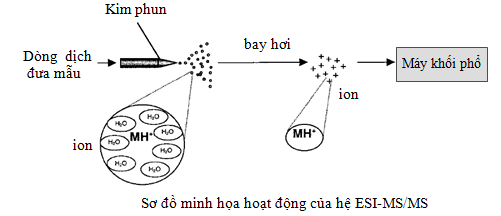
Ngược với MALDI, nguồn ESI ion hóa mẫu trong dịch lỏng. Khi đó peptid sẽ tồn tại ở dạng ion bởi vì chúng chứa các nhóm chức, và điện tích của chúng phụ thuộc vào pH của dung môi. Ở giá trị pH axit (pH 3,5 hoặc thấp hơn) protein/peptid sẽ mang điện tích dương và được phân tách bằng sắc ký nano đa chiều tốt hơn [3]. Do đó, nguồn ESI hay được kết nối trực tiếp với hệ sắc ký lỏng và thường phân tích các peptid ở chế độ ion dương. Hệ ESI-MS/MS có ưu điểm là tự động, ion hóa cao, có khả năng kết nối linh động với sắc ký và khối phổ, peptid thường ở trạng thái đa điện tích (+2, +3...). Trạng thái tích đa điện tích làm cho giá trị m/z phù hợp với giải khối cho phép của các bộ phận phân tích khối như quadrupole và ion-trap. Hệ nanoLC-MS/MS có thể phân tách hỗn hợp protein phức tạp với hàng ngàn protein được nhận dạng.
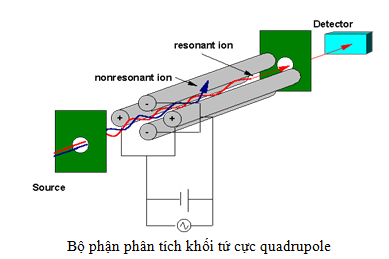
Có ba loại bộ phận phân tách khối liên tục MS/MS (tandem mass analyzer) là triple quadrupole (hình 12), ion-trap và quadrupole-time of flight.
Mặc dù các bộ phận phân tích khối này hoạt động khác nhau nhưng chúng có chung một chức năng là phân tách khối các ion trong điện trường. Hỗn hợp ion được hình thành từ nguồn ESI sẽ được đi qua bộ phận phân tách khối liên tục để phân tách, lựa chọn và vào buồng phân mảnh CID (collision-induced dissociation).
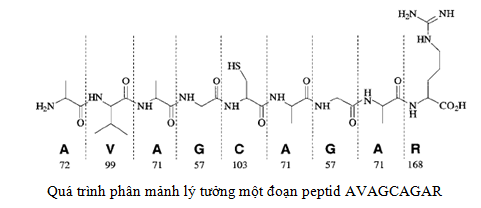
Ion peptid sẽ bị phân mảnh thành các đoạn nhỏ hơn được đo và ghi phổ tại detector. Quá trình phân mảnh liên tục được gọi là quá trình thực hiện MS/MS.
Các phần mềm tin sinh học
Công cụ proteomics quan trọng thứ ba là các phần mềm tin sinh học. Các phần mềm này có thể so sánh, tìm kiếm và hỗ trợ kết quả thu được trên các cơ sở dữ liệu. Các phần mềm ngày nay phát triển rất mạnh, đây là lĩnh vực tin sinh học mới mẻ và hấp dẫn. Những dữ liệu thực nghiệm thu được thực sự cần phân tích bổ sung bằng các phép thử và thuật toán trên máy tính [55]. Do đó từ sự phát triển các phần mềm phân tích, so sánh hình ảnh gel 2 chiều như PD Quest, Quantity one (Bio-Rad), Progenesis (Nonlinear Dynamics) đến các dữ liệu phân tích về khối phổ, và nhiều công cụ đã được bán cùng với các thiết bị tương ứng như MASCOT (Matrix Science) hay SEQUEST (Thermo Finnigan). Các phần mềm này được dùng để so sánh phổ MS/MS thực nghiệm thu được với phổ lý thuyết từ trình tự các protein trên cơ sở dữ liệu (Swiss-Prot, EST sequence data, Genomics sequence data).
Một số công cụ trực tuyến trên Internet qua những liên kết được cung cấp từ ExPASy (www.expasy.ch/tools.html) [56]. Hiện nay, các phần mềm dự đoán cấu trúc, chức năng và vị trí của protein cũng phát triển rất mạnh như QuickGO (http://www.ebi.ac.uk/ego/index.html), amiGO (http://www.geneontology.org/). Những công cụ này cho phép không chỉ nhận diện, mà còn xác định những đặc tính, từ các thuộc tính lý-hóa cơ bản đến dự đoán những cải biến và cả những cấu trúc ba chiều của protein. Những cơ sở dữ liệu về các protein được chú giải, cũng như về hình ảnh điện di hai chiều là điểm cốt lõi của bioinformatics trong nghiên cứu proteome.
Các cơ sở dữ liệu về trình tự gen và protein
Proteomics - khoa học về hệ protein được kế thừa, phát triển trên cơ sở những thành tựu đã đạt được của genomics, và chủ yếu dựa trên các cơ sở dữ liệu về trình tự gen và protein. Ngoài ra, sự phát triển của các kỹ thuật mới như điện di hai chiều (2-DE), sắc ký lỏng nano đa chiều (nanoLC), khối phổ (MS) cũng đang tạo ra một số lượng khổng lồ các số liệu. Tất cả các dữ liệu nói trên đang được xử lý, hợp nhất và cho phép dễ dàng sử dụng cho việc tìm kiếm.
Hiện nay các cơ sở dữ liệu lớn như NCBI (Mỹ), Swiss-Prot (Thụy Sĩ), Gene Ontology Annotation, European Bioinformatics Institute-EBI (Châu Âu), HUPO (Human Proteome Organization) (đa quốc gia), HPP (Human Plasma Proteome), PDB (Protein Data Bank) đã và đang phát triển vô cùng nhanh chóng. Lượng thông tin phong phú do các nghiên cứu về proteome đem lại hoàn toàn bổ trợ cho những thông tin di truyền từ những nghiên cứu về genome. Proteomics sẽ là cơ sở cho sự phát triển của genomics chức năng. Sự phối hợp giữa genomics, proteomics và bioinformatics sẽ đóng vai trò chủ đạo trong các nghiên cứu về sinh-y học, là nền tảng cho sự phát triển các sản phẩm chẩn đoán và chữa trị trong tương lai.